

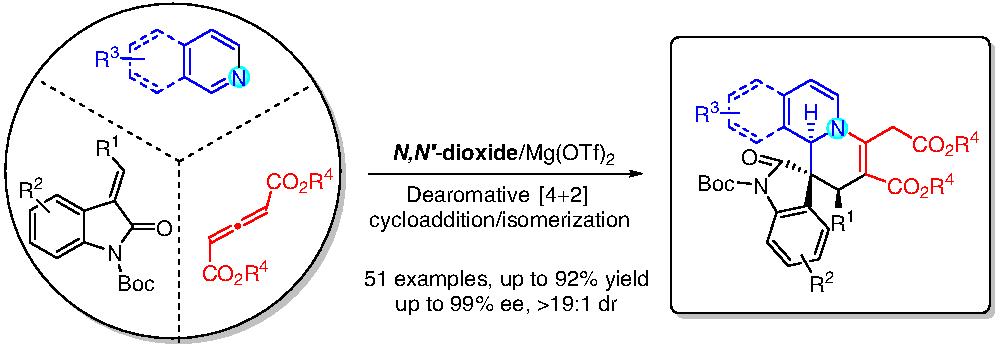
含氮芳香族化合物的催化不对称去芳构化是直接构建手性含氮稠环或螺环化合物的重要方法(Chem. Soc. Rev.2020, 49, 8721–8748)。过去几十年,由含氮芳环(如喹啉、异喹啉、吡啶)与亲电试剂(如炔酸酯、联烯酸酯、重氮酯)发生亲核加成反应,原位生成的Huisgen 1,4-偶极子被广泛应用于合成各种含氮杂环化合物。然而,该类偶极子反应活性高,寿命短,导致反应体系比较复杂,且会产生强的背景反应,其立体选择性控制存在很大挑战,相关的催化不对称反应一直未被实现。
四川大学冯小明课题组发展的手性双氮氧-酰胺化合物(冯配体)是一类优势手性配体,与金属配位形成的手性路易斯酸催化剂(冯催化剂),能够高效高选择性地催化60多类不对称反应。受之前他们课题组关于不对称催化去芳构化研究工作的启发(Angew. Chem. Int. Ed. 2018, 57, 12323–12327; Nat. Commun. 2019, 10, 2116–2125),作者近期报道了首例手性双氮氧-镁(II)配合物催化吲哚酮烯酯、联烯酸酯与含氮芳香杂环的不对称多组分串联反应(图1)。在温和的反应条件下,实现了多种含氮芳香杂环(异喹啉、喹啉、吡啶、啡啶、酞嗪等)的不对称去芳构化,高对映和高非对映选择性地得到了一系列光学活性的含氮多环化合物。
图1手性双氮氧-镁配合物催化不对称去芳构化多组分反应
通过条件筛选,作者确立了以L3-PicH/Mg(OTf)2配合物(1:1,10 mol%)为催化剂,CH2Cl2为溶剂,H2O和Et3N为添加剂的最 优条件。作者首先考察了吲哚酮烯酯的底物范围(图2),结果表明:连接烯烃的酯基对反应的影响较小,不同位阻大小取代的吲哚酮烯酯均能够以高收率和高选择性地转化为相应的二氢异喹啉类产物,即使将酯基更换为杂环(1h-1i)或者苯甲酰基(1j-1l),也能得到不错的结果。相比之下,吲哚酮烯酯苯环上的取代基对反应的对映选择性有较大影响,当连接吸电子取代基时,对映选择性都有不同程度的下降(4r-4t)。随后,作者考察了不同取代的异喹啉,发现取代基的电子效应几乎不影响反应的对映选择性(4u-4ad)。
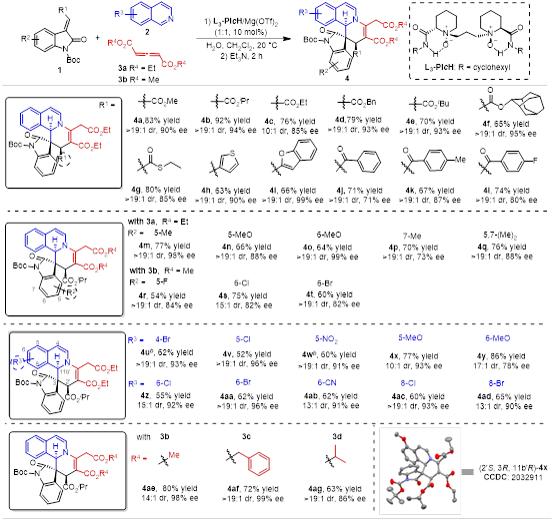
图2不对称去芳构化合成二氢异喹啉类衍生物
吡啶类化合物由于其共振稳定性的增加以及毒化金属催化剂的可能性,使得其参与的催化不对称去芳构化反应比异喹啉类底物更具有挑战性。作者通过改变手性双氮氧配体的结构(L3-PrEt2Me)和延长反应时间,确立了吡啶类底物的最 优反应条件。在此条件下,作者考察了不同取代的吡啶(图3),反应结果表明,含有弱供电子取代基的吡啶类底物,其立体选择性控制并不是很好(6d,6e)。当使用邻位不含取代基的非对称的吡啶时,反应存在区域选择性,受取代基的电子效应和位阻效应双重影响。
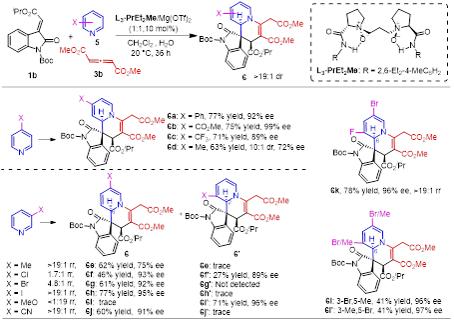
图3不对称去芳构化合成二氢吡啶类衍生物
作者也考察了其它含氮芳香杂环(喹啉、啡啶、酞嗪),以中等到优 秀的对映选择性得到了相应的去芳构化产物,但收率较低(图4)。
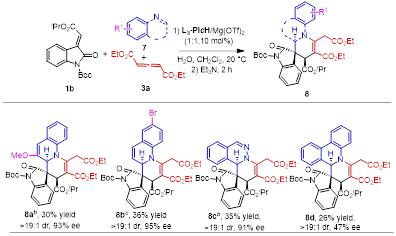
图4其他手性多环含氮杂环的合成
最 后,作者做了一些控制实验来阐明可能的反应机理和添加剂的作用(图5)。包括:1)氘代实验表明产物的生成是经历了分子内的[1,3]-氢迁移(图5a);2)[1,3]-氢迁移进行缓慢,可以通过加入添加剂(Et3N)促进该过程或延长反应时间得到含环内双键的产物。此外,根据之前的文献报道以及产物的绝 对构型,作者也提出了反应可能的历程和过渡态(图5c)。
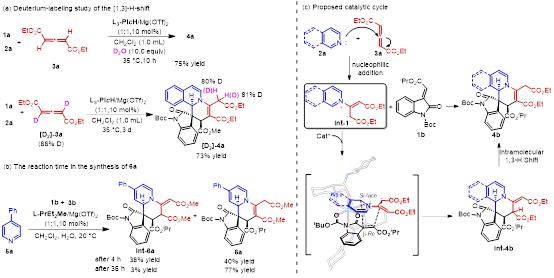
图5控制实验及反应历程
综上所述,该研究工作发展了一种手性双氮氧-镁配合物(冯催化剂)催化的不对称亲核加成/[4+2]环加成去芳构化/异构化多组分串联反应,具有良好的底物普适性和官能团兼容性,高效高选择性地构建了多类光学活性的含氮杂环化合物。该工作以Research Article的形式发表在CCS Chemistry,已在官网“Just Published”栏目上线。
文章详情:
Synthesis of Dihydroisoquinoline and Dihydropyridine Derivatives via Asymmetric Dearomative Three-Component Reaction
Guihua Pan, Changli He, Min Chen, Qian Xiong, Weidi Cao* and Xiaoming Feng*
Citation:CCS Chem. 2021, 3, 2012–2020


